Enzyme mix (red cap)
2 units/μl polymerase I, 0.02 units/μl Dnase I in storage buffer
NT labeling buffer (green cap)
10x conc.
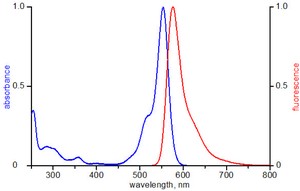
Atto550 NT labeling mix (purple cap)
0.5 mM dATP, 0.5 mM dCTP, 0.5 mM dGTP, 0.25 mM dTTP,
0.25 mM Atto550-dUTP, pH 7.5
Stop buffer (yellow cap)
0.5 M EDTA, pH 8.0
PCR-grade water (white cap)
Recommended NT assay:
Sample Material can be supercoiled or linearized plasmid DNA, cosmid or BAC DNA, whole or partial chromosomes or purified PCR products.
Prepare the following reaction mixture in a sterile vial.
20 μl Nick translation labeling assay
| fill up to 20 μl | PCR-grade water | white cap |
| 2 μl | 10x NT labeling buffer | green cap |
| 2 μl | Atto550 NT
labeling mix | purple cap |
| 1-1.5 μg | template DNA | - |
| 2 μl | Enzyme mix | red cap |
- Vortex the mix gently to assure homogeneity and centrifuge briefly to collect the reaction mixture at the bottom of the tube.
- Place the tube in a precooled thermomixer at 15 °C. An incubation of 90 min is recommended to generate DNA fragments in a size range between 200 and 500 bp.
- To control the length of the fragments load 2 μl of the assay on an agarose gel. Place the reaction tube at -20 °C while running the gel.
- To get smaller fragments add additional 2 μl of the Enzyme mix and extend the incubation at 15 °C.
- For final stopping the reaction add 5 μl of Stop buffer (yellow cap). Proceed to purification or store at -20 °C.
Purification of the probe:
To remove unincorporated nucleotides from the reaction mixture prior to its use in subsequent experiments one of the following procedures is recommended:
1. Purification by silica-gel membrane adsorption - PCR Purification Kit, Cat.-No. PP-201
The Jena Bioscience PCR Purification Kit provides a simple and efficient way to purify DNA fragments larger than 100 bp. The preparation is based on a silica-membrane technology for binding DNA in high-salt and elution in low-salt buffer. Please refer to the instruction manual.
2. Purification by Isopropanol precipitation
Add 1 μl glycogene (2 mg/ml), 2 μl sodium acetate (3 M) and 14 μl isopropanol to the reaction mixture and mix well but gently. Incubate on RT for 15 min and spin down at maximum speed at 4 °C for 30 min. Discard the supernatant and wash 2x with 70 % ethanol (spin down at maximum speed for 5 min).
3. Purification by Centrifugal Filter Units
Unincorporated nucleotides can be removed by centrifugation using centrifugal filter units. Select the filter unit by its cut-off for DNA fragments and follow the manufacturer's instructions.
Incorporation rate of the fluorophore:
The efficiency of DNA labeling can be estimated by calculating the ratio of incorporated fluorophores to the number of bases in the fragment (dye / base).
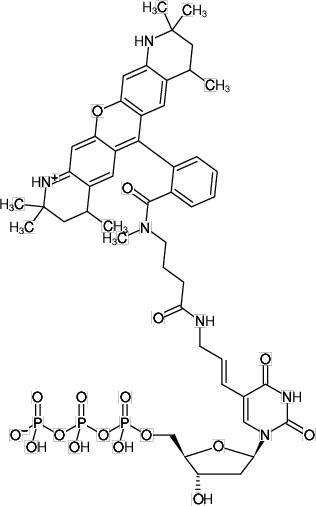
1. Measurement of the optical density: Measure the absorbance of the labeled DNA fragment at 260 nm (A260) and at the excitation maximum (λexc) for the dye (Adye).
2. Correction of the A260 reading: To obtain an accurate absorbance measurement for the nucleic acid, the contribution of the dye at 260 nm has to be corrected. Use the following equation:
Abase = A260 - (Adye x CF260)
Correction Factor for Atto550: CF260 = 0.24
3. Calculation of the labeling rate: The dye to base ratio is given by:
dye / base = (Adye x εbase) / (Abase x εdye)
Extinction coefficients:
Atto550: εdye = 120,000 cm-1 M-1
dsDNA: εbase = 6,600 cm-1 M-1
ssDNA: εbase = 8,900 cm-1 M-1
oligonucleotide: εbase = 10,000 cm-1 M-1
Example: A dye to base ratio of 0.05 corresponds to an incorporation of 10 dye-dUTP nucleotides into a DNA fragment containing 200 nucleotides or, respectively, into a 100 bp PCR fragment. If an equal distribution of dATP, dCTP, dGTP and dTTP in the DNA fragment can be assumed, 10 of the 50 existing dTTPs have been substituted by dye-dUTP resulting in a labeling rate of 20 %.
Related products: Aminoallyl-dUTP-ATTO-550, #NU-803-550